Publications
Peer-reviewed papers, preprints, technical reports, and book citations by Noah Flynn.
Selected work spans agentic AI evaluation, multilingual foundation model adaptation, production model reports, and machine learning for drug metabolism and toxicity. For the most current citation graph, see Google Scholar.
2026
- Preprint
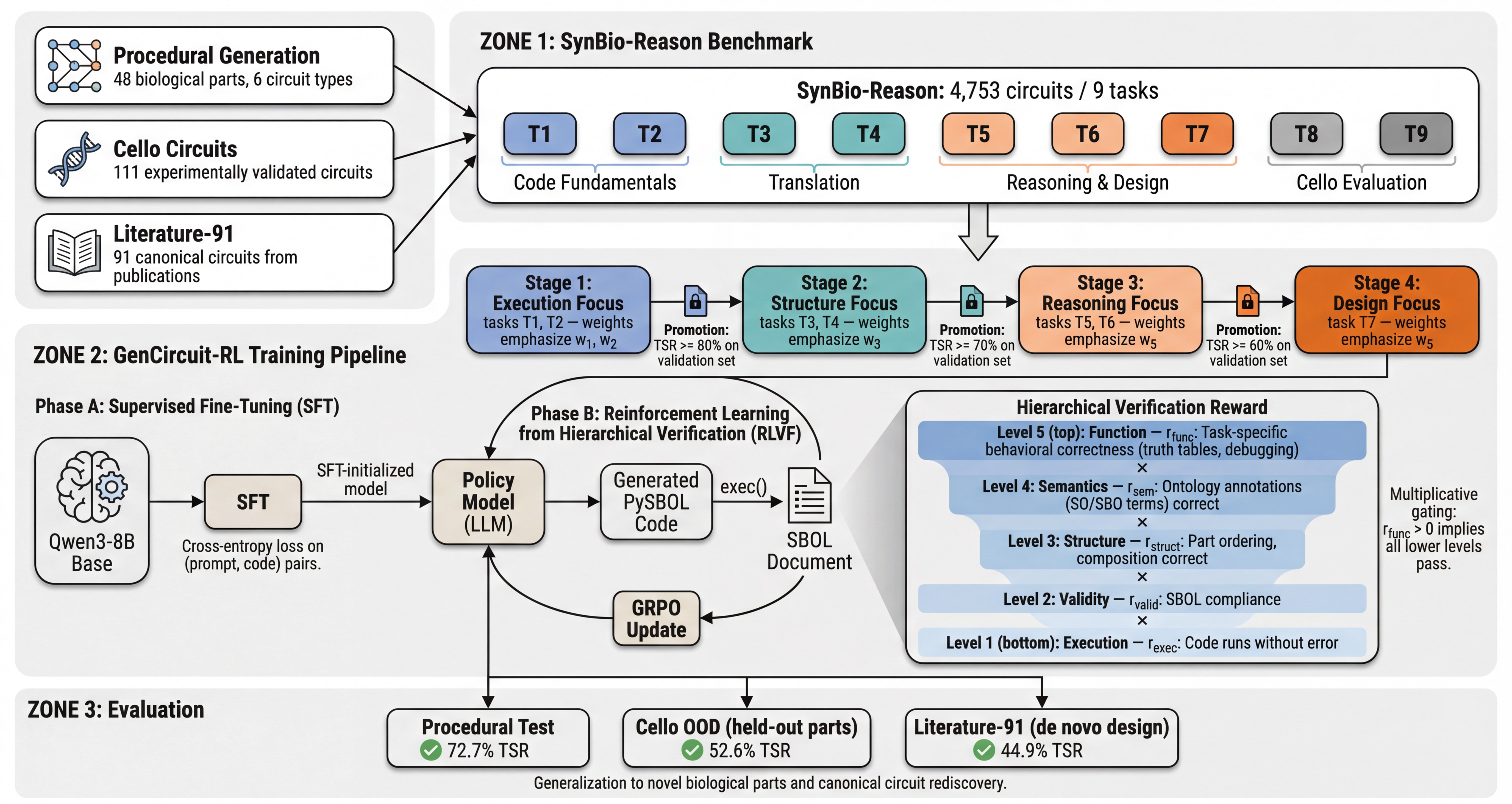 GenCircuit-RL: Reinforcement Learning from Hierarchical Verification for Genetic Circuit DesignNoah R. FlynnICML, 2026
GenCircuit-RL: Reinforcement Learning from Hierarchical Verification for Genetic Circuit DesignNoah R. FlynnICML, 2026@article{flynn2026gencircuit, title = {{GenCircuit-RL}: Reinforcement Learning from Hierarchical Verification for Genetic Circuit Design}, author = {Flynn, Noah R.}, year = {2026}, journal = {ICML}, } - Preprint
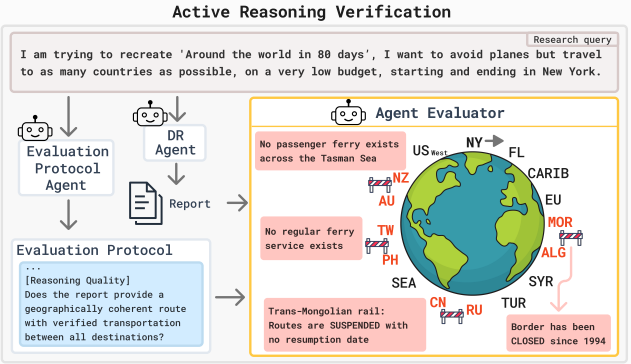 DREAM: Deep Research Evaluation with Agentic MetricsElad Ben Avraham, Changhao Li, Ron Dorfman, and 8 more authorsACL, 2026
DREAM: Deep Research Evaluation with Agentic MetricsElad Ben Avraham, Changhao Li, Ron Dorfman, and 8 more authorsACL, 2026Deep Research Agents generate analyst-grade reports, yet evaluating them remains challenging due to the absence of a single ground truth and the multidimensional nature of research quality. Recent benchmarks propose distinct methodologies, yet they suffer from the Mirage of Synthesis, where strong surface-level fluency and citation alignment can obscure underlying factual and reasoning defects. To address this, we propose DREAM (Deep Research Evaluation with Agentic Metrics), a framework that instantiates the principle of capability parity by making evaluation itself agentic. DREAM structures assessment through an evaluation protocol combining query-agnostic metrics with adaptive metrics generated by a tool-calling agent, enabling temporally aware coverage, grounded verification, and systematic reasoning probes. Controlled evaluations demonstrate DREAM is significantly more sensitive to factual and temporal decay than existing benchmarks, offering a scalable, reference-free evaluation paradigm.
@article{li2026dreambench, title = {{DREAM}: Deep Research Evaluation with Agentic Metrics}, author = {Ben Avraham, Elad and Li, Changhao and Dorfman, Ron and Ganz, Roy and Nuriel, Oren and Dudai, Amir and Aberdam, Aviad and Flynn, Noah R. and Mansimov, Elman and Kalyanpur, Adi and Litman, Ron}, year = {2026}, journal = {ACL}, }
2025
- TMLR
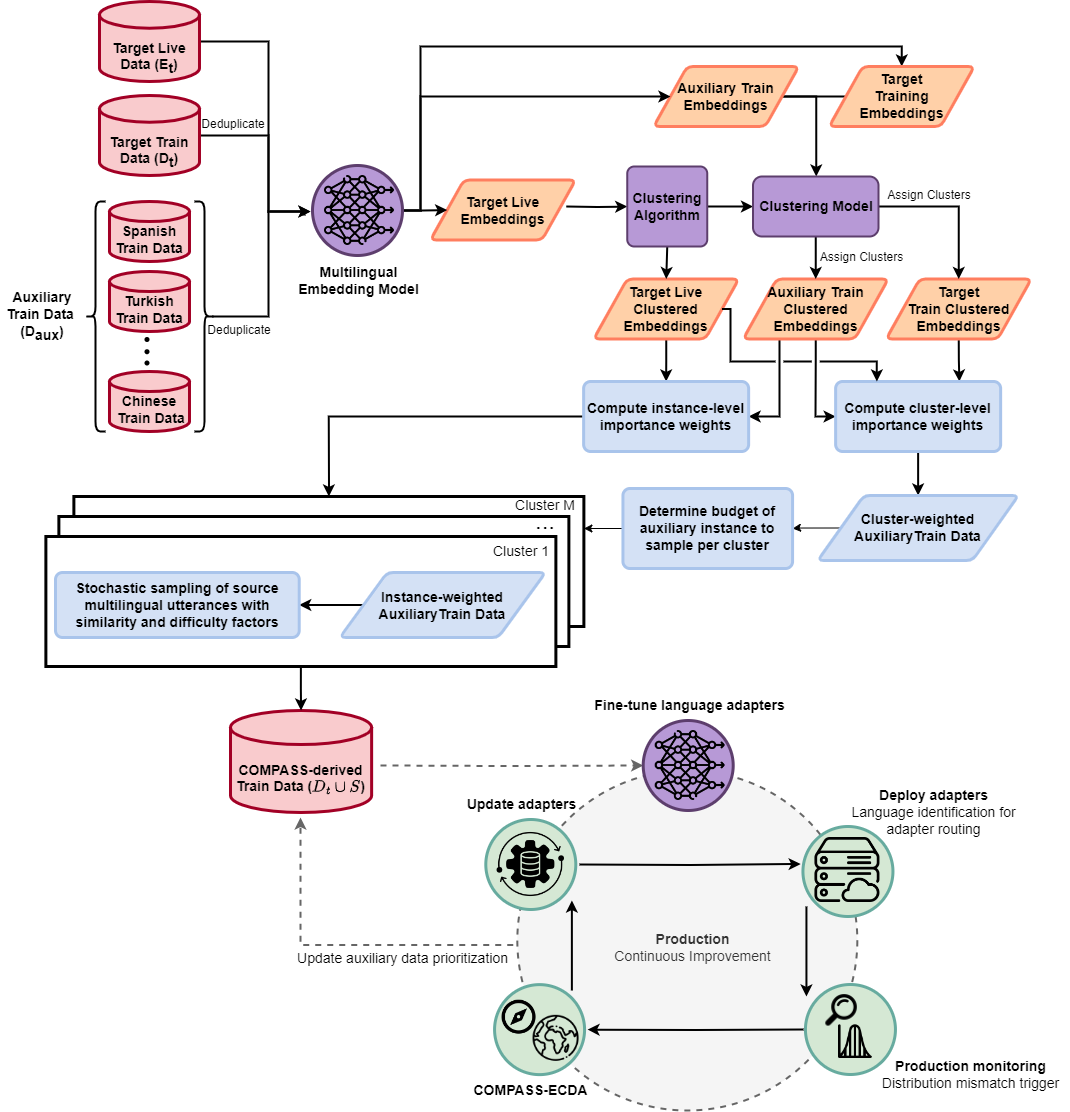 COMPASS: Continual Multilingual PEFT with Adaptive Semantic SamplingNoah R. FlynnTransactions on Machine Learning Research, 2025
COMPASS: Continual Multilingual PEFT with Adaptive Semantic SamplingNoah R. FlynnTransactions on Machine Learning Research, 2025Large language models (LLMs) often exhibit performance disparities across languages, with naive multilingual fine-tuning frequently degrading performance due to negative cross-lingual interference. To address this, we introduce COMPASS (COntinual Multilingual PEFT with Adaptive Semantic Sampling), a novel data-centric framework for adapting LLMs to target languages. COMPASS leverages parameter-efficient fine-tuning (PEFT) by training lightweight, language-specific adapters on a judiciously selected subset of auxiliary multilingual data. The core of our method is a distribution-aware sampling strategy that uses multilingual embeddings and clustering to identify semantic gaps between existing training data and a target usage distribution. By prioritizing auxiliary data from under-represented semantic clusters, COMPASS maximizes positive cross-lingual transfer while minimizing interference. We extend this into a continual learning framework, COMPASS-ECDA, which monitors for data distribution shifts in production and dynamically updates adapters to prevent model staleness, balancing adaptation to new data with the preservation of existing knowledge. Across three different model architectures (Phi-4-Mini, Llama-3.1-8B, and Qwen2.5-7B) and multiple challenging multilingual benchmarks (Global-MMLU, MMLUProX), including unseen long-context tasks (OneRuler), we demonstrate that COMPASS consistently outperforms baseline methods guided by linguistic similarity, providing an effective, efficient, and sustainable solution for developing and maintaining high-performing multilingual models in dynamic environments.
@article{flynn2025compass, title = {{COMPASS}: Continual Multilingual {PEFT} with Adaptive Semantic Sampling}, author = {Flynn, Noah R.}, year = {2025}, journal = {Transactions on Machine Learning Research}, } - NeurIPS
 Case Study in Kinase Inhibitors for Anti-Cancer TherapiesNoah R. FlynnIn NeurIPS Education Track, 2025
Case Study in Kinase Inhibitors for Anti-Cancer TherapiesNoah R. FlynnIn NeurIPS Education Track, 2025This interactive notebook introduces deep learning concepts through a real-world application in pharmaceutical drug discovery. Instead of beginning with abstract mathematical foundations, learners immediately engage with a compelling medical challenge: identifying potential cancer treatments by predicting which molecules might effectively inhibit disease-causing proteins.
@inproceedings{flynn2025kinase, title = {Case Study in Kinase Inhibitors for Anti-Cancer Therapies}, author = {Flynn, Noah R.}, booktitle = {NeurIPS Education Track}, year = {2025}, }

2024
- Amazon
 The Amazon Nova Family of Models: Technical Report and Model CardAmazon AGI, and Noah R.) Flynn2024
The Amazon Nova Family of Models: Technical Report and Model CardAmazon AGI, and Noah R.) Flynn2024We present Amazon Nova, a new generation of state-of-the-art foundation models that deliver frontier intelligence and industry-leading price performance. Amazon Nova Pro is a highly-capable multimodal model with the best combination of accuracy, speed, and cost for a wide range of tasks. Amazon Nova Lite is a low-cost multimodal model that is lightning fast for processing images, video, documents and text. Amazon Nova Micro is a text-only model that delivers our lowest-latency responses at very low cost. Amazon Nova Canvas is an image generation model that creates professional grade images with rich customization controls. Amazon Nova Reel is a video generation model offering high-quality outputs, customization, and motion control. Our models were built responsibly and with a commitment to customer trust, security, and reliability. We report benchmarking results for core capabilities, agentic performance, long context, functional adaptation, runtime performance, and human evaluation.
@techreport{amazonnova2024, title = {The {Amazon Nova} Family of Models: Technical Report and Model Card}, author = {{Amazon AGI} and others (including Flynn, Noah R.)}, year = {2024}, institution = {Amazon Technical Reports}, } - EMNLP
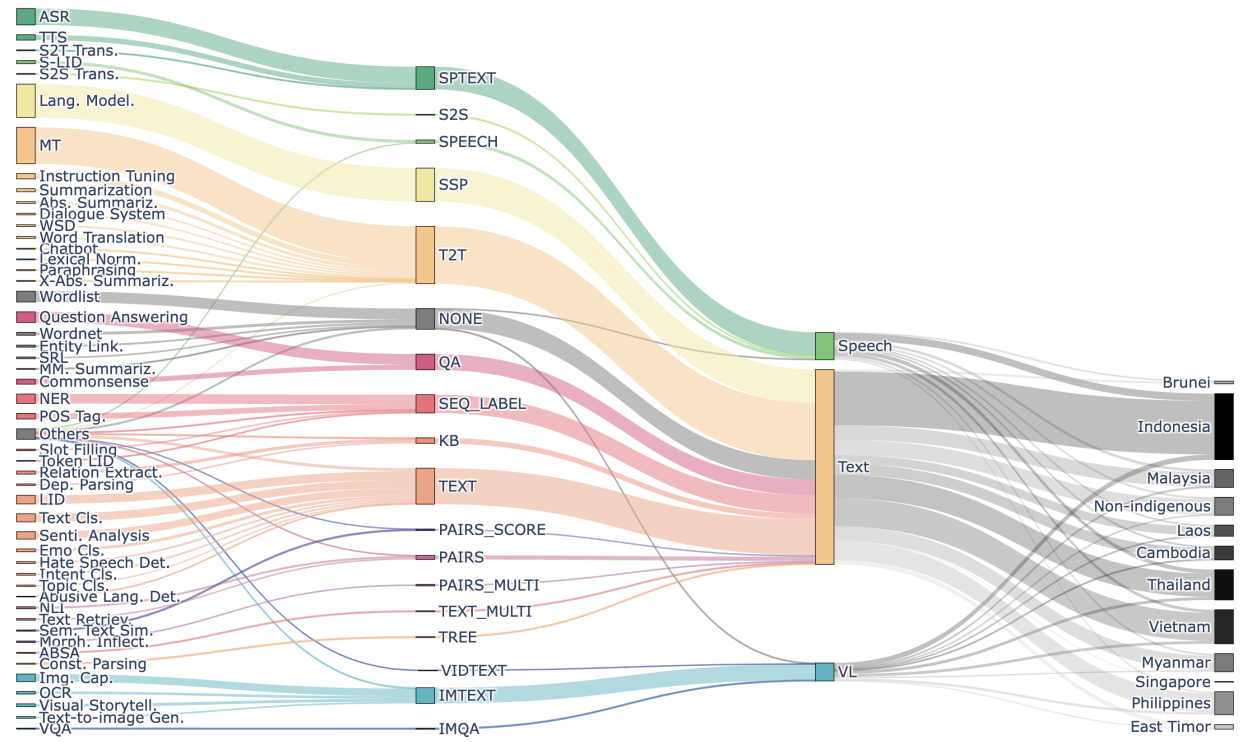 SEACrowd: A Multilingual Multimodal Data Hub and Benchmark Suite for Southeast Asian LanguagesHoly Lovenia, and Noah R.) FlynnIn Proceedings of the 2024 Conference on Empirical Methods in Natural Language Processing (EMNLP), 2024
SEACrowd: A Multilingual Multimodal Data Hub and Benchmark Suite for Southeast Asian LanguagesHoly Lovenia, and Noah R.) FlynnIn Proceedings of the 2024 Conference on Empirical Methods in Natural Language Processing (EMNLP), 2024Southeast Asia (SEA) is a region rich in linguistic diversity and cultural variety, with over 1,300 indigenous languages and a population of 671 million people. However, prevailing AI models suffer from a significant lack of representation of texts, images, and audio datasets from SEA, compromising the quality of AI models for SEA languages. Evaluating models for SEA languages is challenging due to the scarcity of high-quality datasets, compounded by the dominance of English training data, raising concerns about potential cultural misrepresentation. To address these challenges, we introduce SEACrowd, a collaborative initiative that consolidates a comprehensive resource hub that fills the resource gap by providing standardized corpora in nearly 1,000 SEA languages across three modalities. Through our SEACrowd benchmarks, we assess the quality of AI models on 36 indigenous languages across 13 tasks, offering valuable insights into the current AI landscape in SEA. Furthermore, we propose strategies to facilitate greater AI advancements, maximizing potential utility and resource equity for the future of AI in SEA.
@inproceedings{lovenia2024seacrowd, title = {{SEACrowd}: A Multilingual Multimodal Data Hub and Benchmark Suite for Southeast Asian Languages}, author = {Lovenia, Holy and others (including Flynn, Noah R.)}, booktitle = {Proceedings of the 2024 Conference on Empirical Methods in Natural Language Processing (EMNLP)}, year = {2024}, }
2023
- Front. Pharm.
 Editorial: Advancements in Computational Studies of Drug ToxicityNoah R. Flynn, Gary P. Miller, and S. Joshua SwamidassFrontiers in Pharmacology, 2023
Editorial: Advancements in Computational Studies of Drug ToxicityNoah R. Flynn, Gary P. Miller, and S. Joshua SwamidassFrontiers in Pharmacology, 2023@article{flynn2023drugtoxeditorial, title = {Editorial: Advancements in Computational Studies of Drug Toxicity}, author = {Flynn, Noah R. and Miller, Gary P. and Swamidass, S. Joshua}, year = {2023}, journal = {Frontiers in Pharmacology}, doi = {10.3389/fphar.2023.1230409} } - JCIM
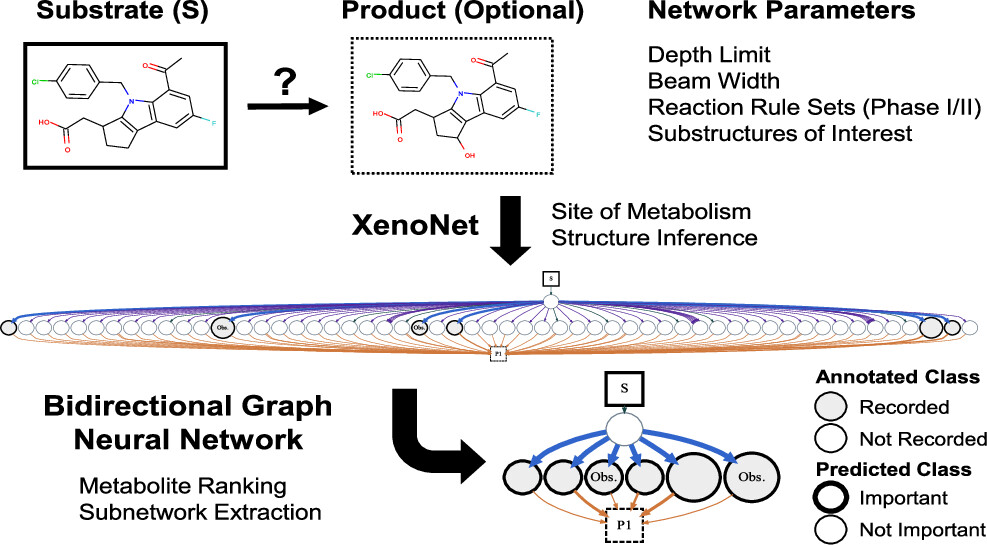 Message Passing Neural Networks Improve Prediction of Metabolite AuthenticityNoah R. Flynn, and S. Joshua SwamidassJournal of Chemical Information and Modeling, 2023
Message Passing Neural Networks Improve Prediction of Metabolite AuthenticityNoah R. Flynn, and S. Joshua SwamidassJournal of Chemical Information and Modeling, 2023Cytochrome P450 enzymes aid in the elimination of a preponderance of small molecule drugs, but can generate reactive metabolites that may adversely react with protein and DNA and prompt drug candidate attrition or market withdrawal. We previously introduced XenoNet, a method to enumerate and predict likely metabolic pathways and intermediate metabolites formed during drug metabolism. Here, we extend XenoNet with a bidirectional message passing neural network that integrates edge feature information and local network structure using edge-conditioned graph convolutions and jumping knowledge to predict the authenticity of inferred Phase I metabolite structures. We test on 311 metabolic networks, containing 6606 intermediates, annotated from 20 736 reaction records derived from chemical literature and Cytochrome P450 databases. Our model achieved area under the receiver operating characteristic curves of 88.5 and 87.6% for separating experimentally observed and unobserved metabolites at global and network levels, respectively. Our approach is robust across varying network sizes, accurately captures metabolites that appear or disappear depending on context, extracts important metabolic subnetworks, and identifies known bioactivation pathways for drugs like nimesulide and terbinafine. Our method can accurately suggest unreported metabolites for experimental study and may rationalize modifications for avoiding deleterious pathways antecedent to reactive metabolite formation.
@article{flynn2023mpnn, title = {Message Passing Neural Networks Improve Prediction of Metabolite Authenticity}, author = {Flynn, Noah R. and Swamidass, S. Joshua}, year = {2023}, journal = {Journal of Chemical Information and Modeling}, }
2022
- Front. Pharm.
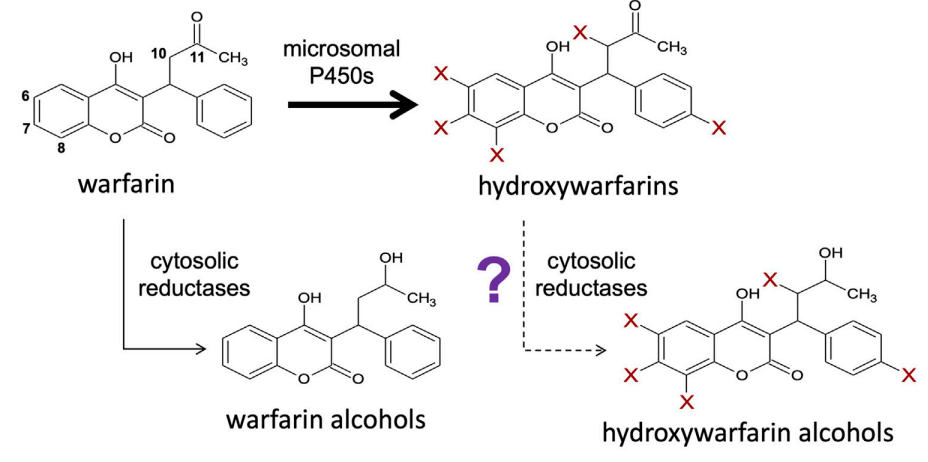 Discovery of Novel Reductive Elimination Pathway for 10-HydroxywarfarinDakota L. Pouncey, and Noah R.) FlynnFrontiers in Pharmacology, 2022
Discovery of Novel Reductive Elimination Pathway for 10-HydroxywarfarinDakota L. Pouncey, and Noah R.) FlynnFrontiers in Pharmacology, 2022Coumadin (R/S-warfarin) anticoagulant therapy is highly efficacious in preventing the formation of blood clots; however, significant inter-individual variations in response risks over or under dosing resulting in adverse bleeding events or ineffective therapy, respectively. Hydroxywarfarin metabolites are more potent anticoagulants than warfarin, yet their elimination mechanism(s) remain largely uncharacterized. We hypothesized that hydroxywarfarins undergo reduction and carried out experimental steady-state reactions with human liver cytosol for conversion of various hydroxywarfarin isomers to corresponding alcohols. CBR1 and to a lesser extent AKR1C3 reductases are responsible for 10-hydroxywarfarin reduction, and due to the inefficiency of reactions, only reduction of 10-hydroxywarfarin is likely to be important in clearance of the metabolite. This pathway for 10-hydroxywarfarin may have clinical relevance given its anticoagulant activity and capacity to inhibit S-warfarin metabolism.
@article{pouncey2022hydroxywarfarin, title = {Discovery of Novel Reductive Elimination Pathway for 10-Hydroxywarfarin}, author = {Pouncey, Dakota L. and others (including Flynn, Noah R.)}, year = {2022}, journal = {Frontiers in Pharmacology}, }
2021
- IJCNN
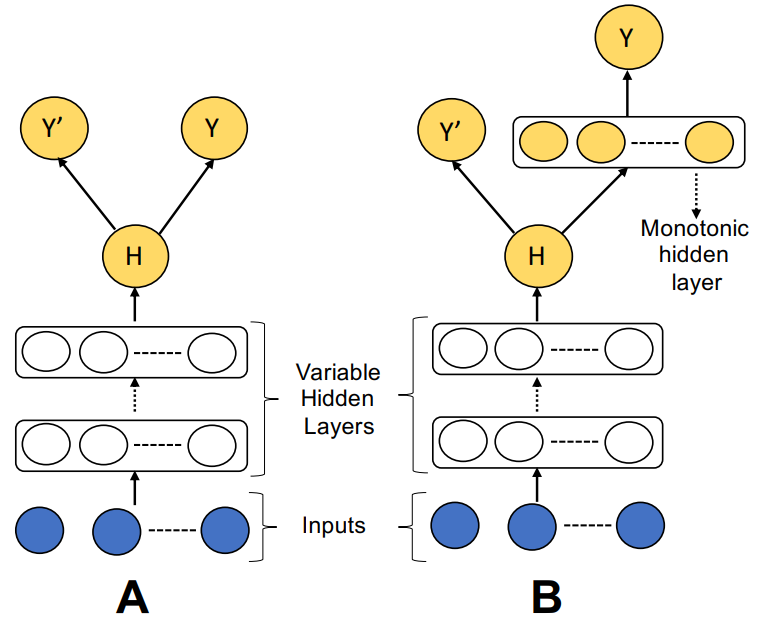 Cal-net: Jointly Learning Classification and Calibration on Imbalanced Binary Classification TasksArghya Datta, Noah R. Flynn, and S. Joshua SwamidassIn International Joint Conference on Neural Networks (IJCNN), 2021
Cal-net: Jointly Learning Classification and Calibration on Imbalanced Binary Classification TasksArghya Datta, Noah R. Flynn, and S. Joshua SwamidassIn International Joint Conference on Neural Networks (IJCNN), 2021Datasets in critical domains are often class imbalanced, with a minority class far rarer than the majority class, and classification models face challenges to produce calibrated predictions on these datasets. A common approach to address this issue is to train classification models in the first step and subsequently use post-processing parametric or non-parametric calibration techniques to re-scale the model’s outputs in the second step without tuning any underlying parameters in the model to improve calibration. However, these common approaches are vulnerable to class imbalanced data, often producing unstable results that do not jointly optimize classification or calibration performance. We propose Cal-Net, a novel self-calibrating neural network architecture that simultaneously optimizes classification and calibration performances for class imbalanced datasets in a single training phase, thereby eliminating the need for any post-processing procedure. Empirical results show that Cal-Net outperforms far more complex neural networks and post-processing calibration techniques in both classification and calibration performances on four synthetic and four benchmark class imbalanced binary classification datasets. Cal-Net can readily be extended to more complicated learning tasks, online learning and can be incorporated in more complex architectures.
@inproceedings{datta2021calnet, title = {Cal-net: Jointly Learning Classification and Calibration on Imbalanced Binary Classification Tasks}, author = {Datta, Arghya and Flynn, Noah R. and Swamidass, S. Joshua}, booktitle = {International Joint Conference on Neural Networks (IJCNN)}, year = {2021}, } - Metabolites
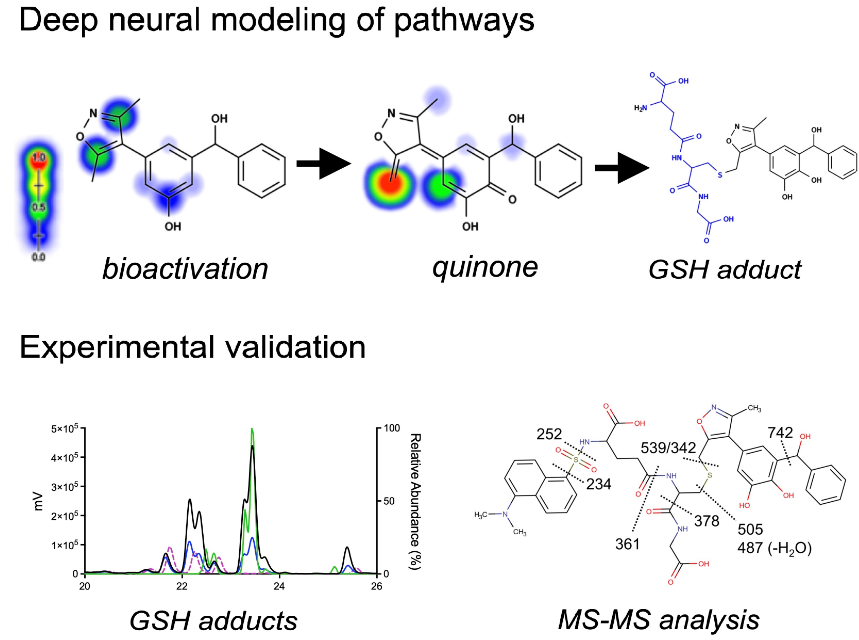 Bioactivation of Isoxazole-Containing Bromodomain and Extra-Terminal Domain (BET) InhibitorsNoah R. Flynn, Michael D. Ward, Mary A. Schleiff, and 1 more authorMetabolites, 2021
Bioactivation of Isoxazole-Containing Bromodomain and Extra-Terminal Domain (BET) InhibitorsNoah R. Flynn, Michael D. Ward, Mary A. Schleiff, and 1 more authorMetabolites, 2021The 3,5-dimethylisoxazole motif has become a useful and popular acetyl-lysine mimic employed in isoxazole-containing bromodomain and extra-terminal (BET) inhibitors but may introduce the potential for bioactivations into toxic reactive metabolites. We coupled deep neural models for quinone formation, metabolite structures, and biomolecule reactivity to predict bioactivation pathways for 32 BET inhibitors and validate the bioactivation of select inhibitors experimentally. Subsequent experimental studies confirmed the formation of both types of quinones for OXFBD molecules, yet traditional quinones were the dominant reactive metabolites. The coupled modeling approach predicted BET inhibitor bioactivations including novel extended quinone methides, and experimentally verified those pathways highlighting potential concerns for toxicity in the development of these new drug leads.
@article{flynn2021bromo, title = {Bioactivation of Isoxazole-Containing Bromodomain and Extra-Terminal Domain ({BET}) Inhibitors}, author = {Flynn, Noah R. and Ward, Michael D. and Schleiff, Mary A. and others}, year = {2021}, journal = {Metabolites}, } - PLOS CB
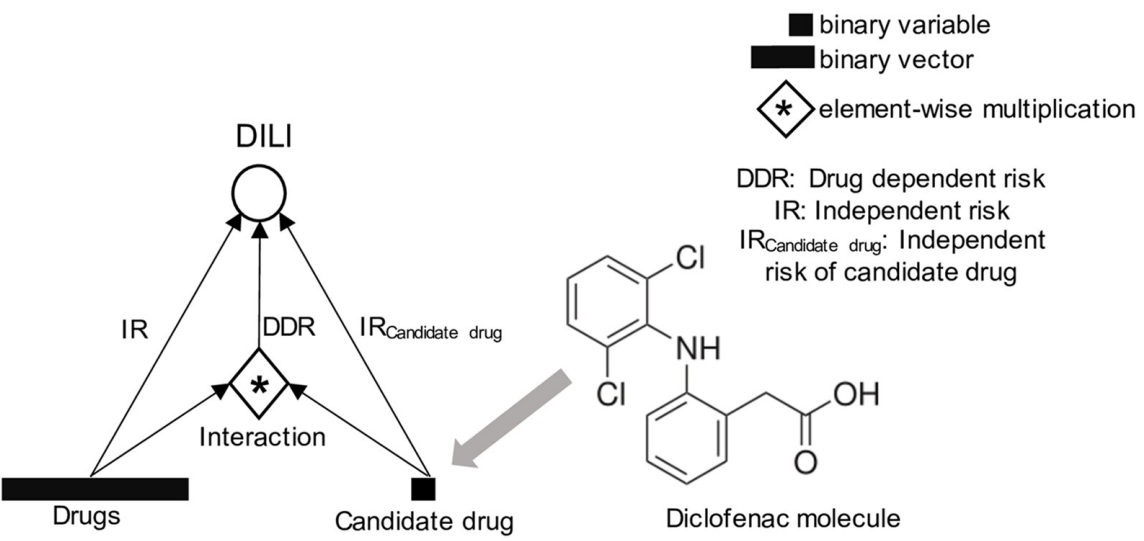 Machine Learning on Liver-Injuring Drug Interactions with NSAIDs from Hospitalization DataArghya Datta‡, Noah R. Flynn‡, Dustyn A. Barnette, and 1 more authorPLOS Computational Biology, 2021‡ Equal contribution
Machine Learning on Liver-Injuring Drug Interactions with NSAIDs from Hospitalization DataArghya Datta‡, Noah R. Flynn‡, Dustyn A. Barnette, and 1 more authorPLOS Computational Biology, 2021‡ Equal contributionDrug-drug interactions account for up to 30% of adverse drug reactions. Increasing prevalence of electronic health records (EHRs) offers a unique opportunity to build machine learning algorithms to identify drug-drug interactions that drive adverse events. In this study, we investigated hospitalizations’ data to study drug interactions with non-steroidal anti-inflammatory drugs (NSAIDS) that result in drug-induced liver injury (DILI). We propose a logistic regression based machine learning algorithm that unearths several known interactions from an EHR dataset of about 400,000 hospitalization. Our proposed modeling framework is successful in detecting 87.5% of the positive controls, which are defined by drugs known to interact with diclofenac causing an increased risk of DILI, and correctly ranks aggregate risk of DILI for eight commonly prescribed NSAIDs. We found that our modeling framework is particularly successful in inferring associations of drug-drug interactions from relatively small EHR datasets. Furthermore, we have identified a novel and potentially hepatotoxic interaction that might occur during concomitant use of meloxicam and esomeprazole, which are commonly prescribed together to allay NSAID-induced gastrointestinal (GI) bleeding. Empirically, we validate our approach against prior methods for signal detection on EHR datasets, in which our proposed approach outperforms all the compared methods across most metrics, such as area under the receiver operating characteristic curve (AUROC) and area under the precision-recall curve (AUPRC).
@article{datta2021ehr, title = {Machine Learning on Liver-Injuring Drug Interactions with {NSAIDs} from Hospitalization Data}, author = {Datta, Arghya and Flynn, Noah R. and Barnette, Dustyn A. and others}, year = {2021}, journal = {PLOS Computational Biology}, note = {‡ Equal contribution} } - Chem. Res. Tox.
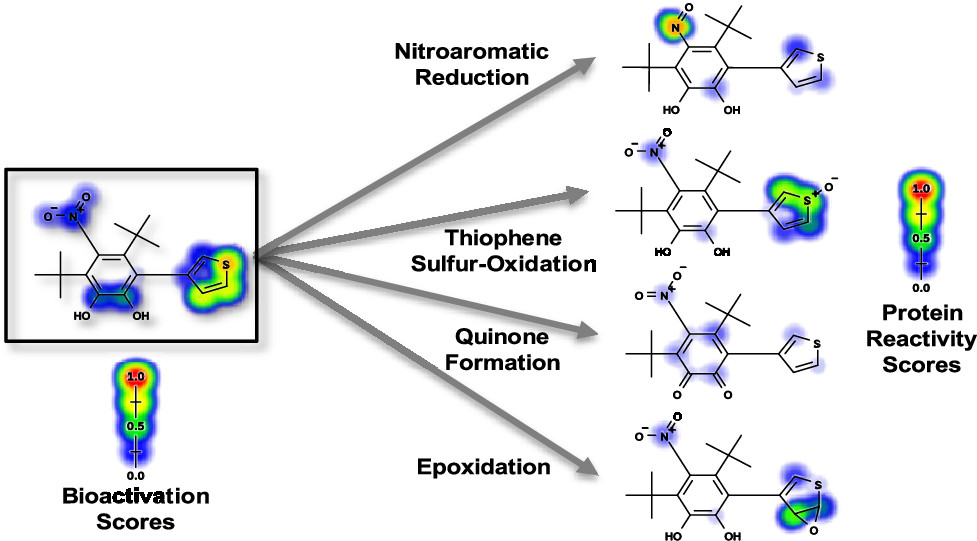 Modeling the Bioactivation and Subsequent Reactivity of DrugsTyler B. Hughes‡, Noah R. Flynn‡, Na Le Dang, and 1 more authorChemical Research in Toxicology, 2021‡ Equal contribution
Modeling the Bioactivation and Subsequent Reactivity of DrugsTyler B. Hughes‡, Noah R. Flynn‡, Na Le Dang, and 1 more authorChemical Research in Toxicology, 2021‡ Equal contributionElectrophilically reactive drug metabolites are implicated in many adverse drug reactions. We modeled four of the most common metabolic transformations that result in bioactivation: quinone formation, epoxidation, thiophene sulfur-oxidation, and nitroaromatic reduction. We synthesized models of metabolism and reactivity, combining them in a feedforward neural network to generate bioactivation predictions at both the pathway and molecule level. Among molecules bioactivated by these pathways, the model predicted the correct pathway with an AUC accuracy of 89.98%, and predicted whether molecules will be bioactivated, distinguishing bioactivated and nonbioactivated molecules with 81.06% AUC. The known bioactivation pathways of alclofenac and benzbromarone were identified by the algorithm, and high probability bioactivation pathways not yet confirmed were identified for safrazine, zimelidine, and astemizole. This computational approach enables drug candidates to be quickly evaluated for a toxicity risk that often evades detection during preclinical trials.
@article{hughes2021bioactivation, title = {Modeling the Bioactivation and Subsequent Reactivity of Drugs}, author = {Hughes, Tyler B. and Flynn, Noah R. and Dang, Na Le and Swamidass, S. Joshua}, year = {2021}, journal = {Chemical Research in Toxicology}, note = {‡ Equal contribution} } - DMD
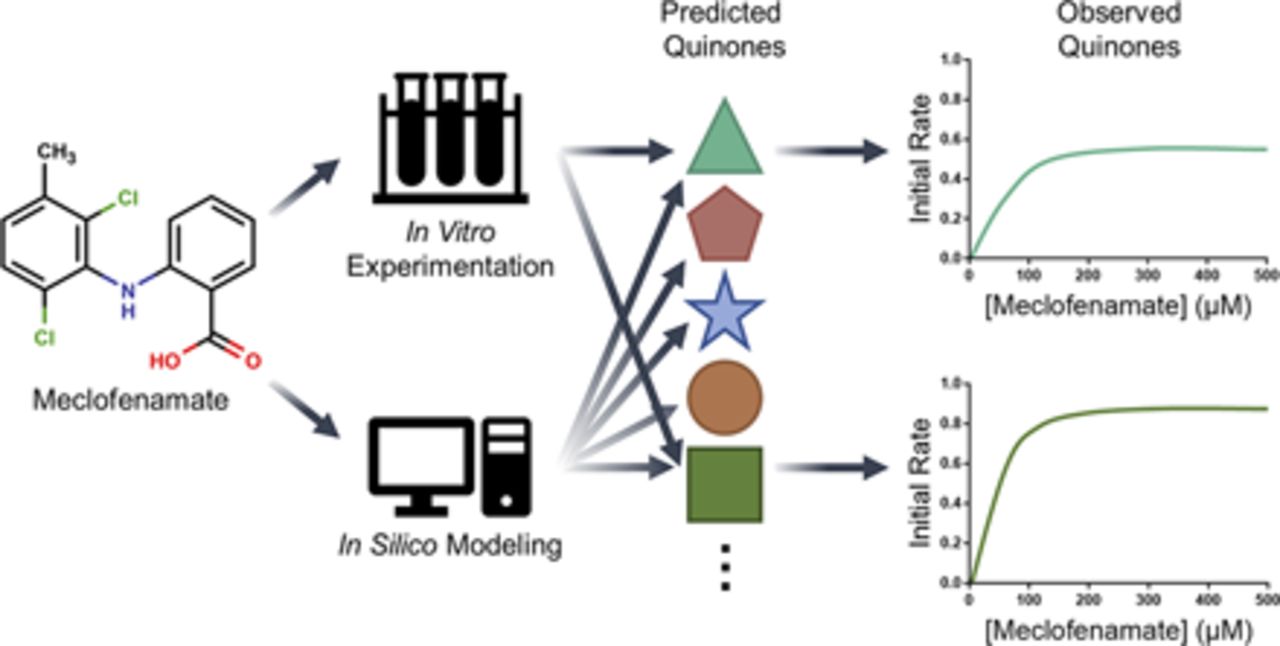 Significance of Multiple Bioactivation Pathways for Meclofenamate as Revealed through Modeling and Reaction KineticsMary A. Schleiff, Noah R. Flynn, Sasin Payakachat, and 1 more authorDrug Metabolism and Disposition, 2021
Significance of Multiple Bioactivation Pathways for Meclofenamate as Revealed through Modeling and Reaction KineticsMary A. Schleiff, Noah R. Flynn, Sasin Payakachat, and 1 more authorDrug Metabolism and Disposition, 2021Meclofenamate is a nonsteroidal anti-inflammatory drug used in the treatment of mild-to-moderate pain yet poses a rare risk of hepatotoxicity through an unknown mechanism. Nonsteroidal anti-inflammatory drug (NSAID) bioactivation is a common molecular initiating event for hepatotoxicity. Thus, we hypothesized a similar mechanism for meclofenamate and leveraged computational and experimental approaches to identify and characterize its bioactivation. Analyses employing our XenoNet model indicated possible pathways to meclofenamate bioactivation into 19 reactive metabolites subsequently trapped into glutathione adducts. We describe the first reported bioactivation kinetics for meclofenamate and relative importance of those pathways using human liver microsomes. The findings validated only four of the many bioactivation pathways predicted by modeling. For experimental studies, dansyl glutathione was a critical trap for reactive quinone metabolites and provided a way to characterize adduct structures by mass spectrometry and quantitate yields during reactions. Of the four quinone adducts, we were able to characterize structures for three of them. Based on kinetics, the most efficient bioactivation pathway led to the monohydroxy para-quinone-imine followed by the dechloro-ortho-quinone-imine. Two very inefficient pathways led to the dihydroxy ortho-quinone and a likely multiply adducted quinone. When taken together, bioactivation pathways for meclofenamate accounted for approximately 13% of total metabolism.
@article{schleiff2021meclofenamate, title = {Significance of Multiple Bioactivation Pathways for Meclofenamate as Revealed through Modeling and Reaction Kinetics}, author = {Schleiff, Mary A. and Flynn, Noah R. and Payakachat, Sasin and others}, year = {2021}, journal = {Drug Metabolism and Disposition}, }
2020
- Tox. Lett.
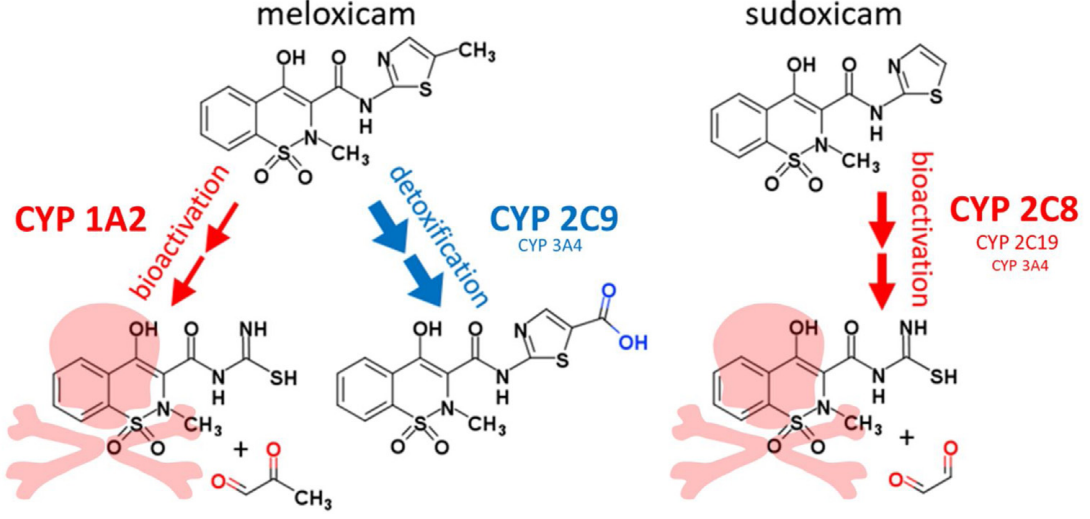 Meloxicam Methyl Group Determines Enzyme Specificity for Thiazole Bioactivation Compared to SudoxicamDustyn A. Barnette, and Noah R.) FlynnToxicology Letters, 2020
Meloxicam Methyl Group Determines Enzyme Specificity for Thiazole Bioactivation Compared to SudoxicamDustyn A. Barnette, and Noah R.) FlynnToxicology Letters, 2020Meloxicam is a thiazole-containing NSAID that was approved for marketing with favorable clinical outcomes despite being structurally similar to the hepatotoxic sudoxicam. Introduction of a single methyl group on the thiazole results in an overall lower toxic risk, yet the group’s impact on P450 isozyme bioactivation is unclear. Through analytical methods, we used inhibitor phenotyping and recombinant P450s to identify contributing P450s, and then measured steady-state kinetics for bioactivation of sudoxicam and meloxicam by the recombinant P450s to determine relative efficiencies. Experiments showed that CYP2C8, 2C19, and 3A4 catalyze sudoxicam bioactivation, and CYP1A2 catalyzes meloxicam bioactivation, indicating that the methyl group not only impacts enzyme affinity for the drugs, but also alters which isozymes catalyze the metabolic pathways. Scaling of relative P450 efficiencies based on average liver concentration revealed that CYP2C8 dominates the sudoxicam bioactivation pathway and CYP2C9 dominates meloxicam detoxification. Dominant P450s were applied for an informatics assessment of electronic health records to identify potential correlations between meloxicam drug-drug interactions and drug-induced liver injury. Overall, our findings provide a cautionary tale on assumed impacts of even simple structural modifications on drug bioactivation while also revealing specific targets for clinical investigations of predictive factors that determine meloxicam-induced idiosyncratic liver injury.
@article{barnette2020meloxicam, title = {Meloxicam Methyl Group Determines Enzyme Specificity for Thiazole Bioactivation Compared to Sudoxicam}, author = {Barnette, Dustyn A. and others (including Flynn, Noah R.)}, year = {2020}, journal = {Toxicology Letters}, } - JCIM
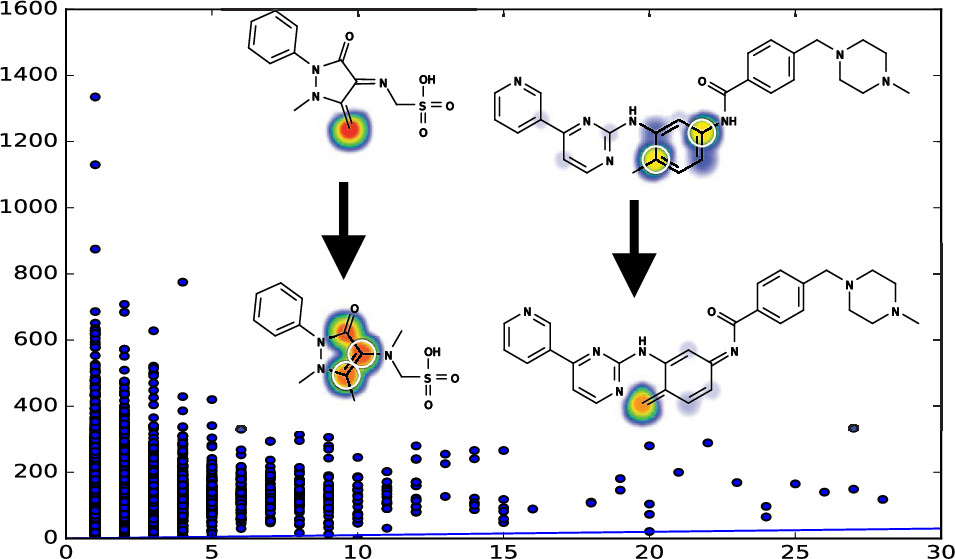 The Metabolic Forest: Predicting the Diverse Structures of Drug MetabolitesTyler B. Hughes, Na Le Dang, Ayush Kumar, and 2 more authorsJournal of Chemical Information and Modeling, 2020
The Metabolic Forest: Predicting the Diverse Structures of Drug MetabolitesTyler B. Hughes, Na Le Dang, Ayush Kumar, and 2 more authorsJournal of Chemical Information and Modeling, 2020Adverse drug metabolism often severely impacts patient morbidity and mortality. Unfortunately, drug metabolism experimental assays are costly, inefficient, and slow. Instead, computational modeling could rapidly flag potentially toxic molecules across thousands of candidates in the early stages of drug development. Most metabolism models focus on predicting sites of metabolism (SOMs): the specific substrate atoms targeted by metabolic enzymes. However, SOMs are merely a proxy for metabolic structures: knowledge of an SOM does not explicitly provide the actual metabolite structure. Without an explicit metabolite structure, computational systems cannot evaluate the new molecule’s properties. For example, the metabolite’s reactivity cannot be automatically predicted, a crucial limitation because reactive drug metabolites are a key driver of adverse drug reactions (ADRs). Additionally, further metabolic events cannot be forecast, even though the metabolic path of the majority of substrates includes two or more sequential steps. To overcome the myopia of the SOM paradigm, this study constructs a well-defined system—termed the metabolic forest—for generating exact metabolite structures. We validate the metabolic forest with the substrate and product structures from a large, chemically diverse, literature-derived dataset of 20 736 records. The metabolic forest finds a pathway linking each substrate and product for 79.42% of these records. By performing a breadth-first search of depth two or three, we improve performance to 88.43 and 88.77%, respectively. The metabolic forest includes a specialized algorithm for producing accurate quinone structures, the most common type of reactive metabolite. To our knowledge, this quinone structure algorithm is the first of its kind, as the diverse mechanisms of quinone formation are difficult to systematically reproduce. We validate the metabolic forest on a previously published dataset of 576 quinone reactions, predicting their structures with a depth three performance of 91.84%. The metabolic forest accurately enumerates metabolite structures, enabling promising new directions such as joint metabolism and reactivity modeling.
@article{hughes2020metforest, title = {The Metabolic Forest: Predicting the Diverse Structures of Drug Metabolites}, author = {Hughes, Tyler B. and Dang, Na Le and Kumar, Ayush and Flynn, Noah R. and Swamidass, S. Joshua}, year = {2020}, journal = {Journal of Chemical Information and Modeling}, } - JCIM
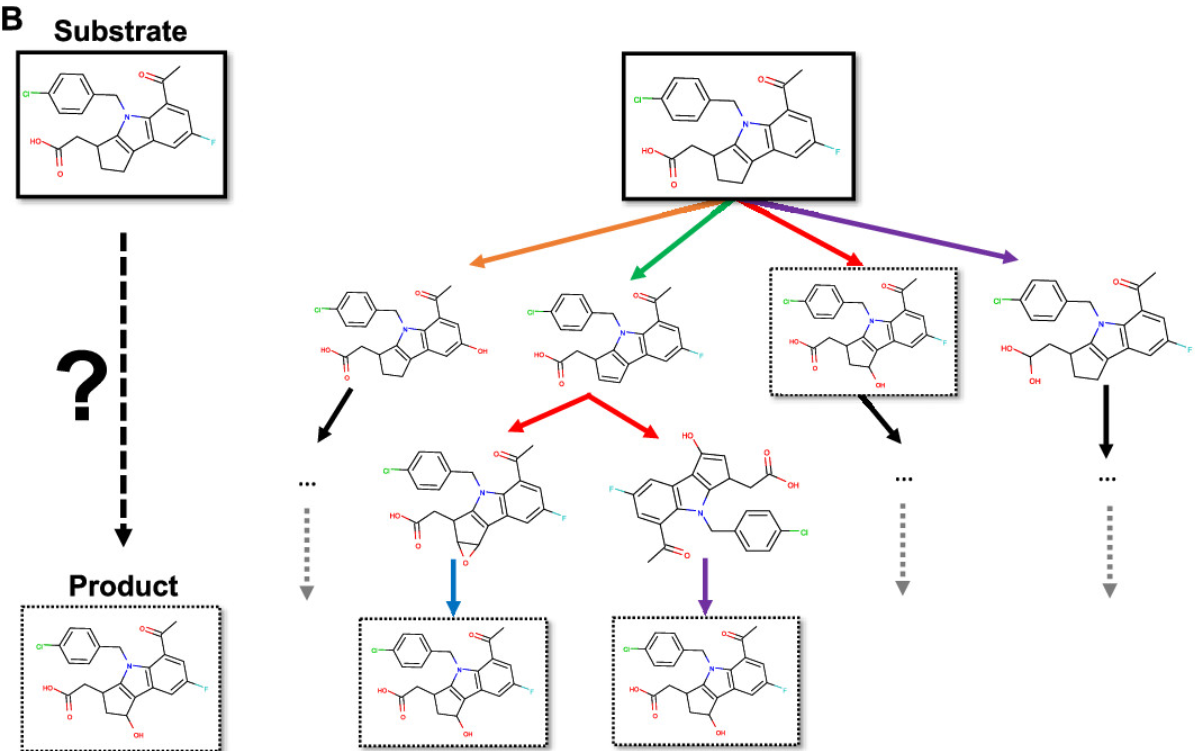 XenoNet: Inference and Likelihood of Intermediate Metabolite FormationNoah R. Flynn, Na Le Dang, Michael D. Ward, and 1 more authorJournal of Chemical Information and Modeling, 2020
XenoNet: Inference and Likelihood of Intermediate Metabolite FormationNoah R. Flynn, Na Le Dang, Michael D. Ward, and 1 more authorJournal of Chemical Information and Modeling, 2020Drug metabolism is a common cause of adverse drug reactions. Drug molecules can be metabolized into reactive metabolites, which can conjugate to biomolecules, like protein and DNA, in a process termed bioactivation. Cytochrome P450 (P450) enzymes metabolize drugs and generate reactive metabolites. However, traditional experimental and computational methods are insufficient to uncover all of the possible intermediate metabolites that form during drug metabolism. We developed XenoNet, a metabolic network predictor that takes a pair of a substrate and target product as input and enumerates pathways, or sequences of intermediate metabolite structures, between the pair. XenoNet then computes the likelihood of those pathways and intermediate metabolites. XenoNet was trained on all publicly available metabolic networks constructed from metabolite structures. We validated XenoNet against 17 054 metabolic networks and found XenoNet can predict experimentally observed pathways and intermediate metabolites linking the input substrate and product pair with a recall of 88 and 46%, respectively. With likelihood scoring, we achieved a top-one pathway and intermediate metabolite accuracy of 93.6 and 51.9%, respectively, and outperformed popular prediction methods including BioTransformer and SyGMa. XenoNet is available at https://swami.wustl.edu/xenonet.
@article{flynn2020xenonet, title = {{XenoNet}: Inference and Likelihood of Intermediate Metabolite Formation}, author = {Flynn, Noah R. and Dang, Na Le and Ward, Michael D. and Swamidass, S. Joshua}, year = {2020}, journal = {Journal of Chemical Information and Modeling}, } - Toxicology
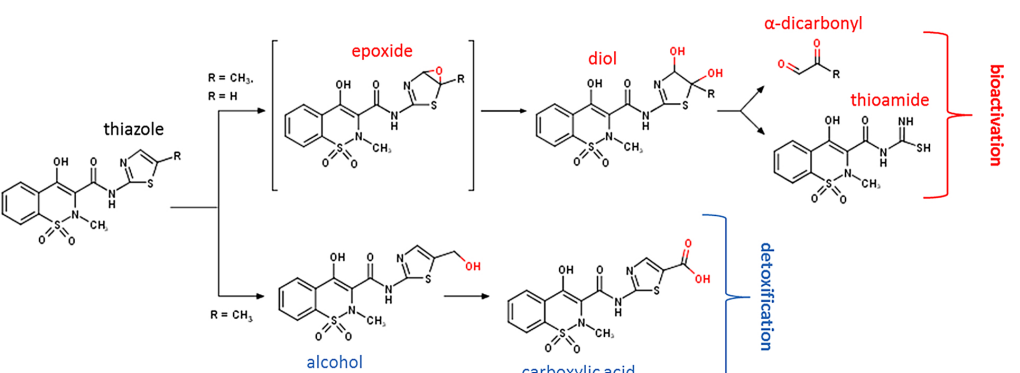 Dual Mechanisms Suppress Meloxicam Bioactivation Relative to SudoxicamDustyn A. Barnette, and Noah R.) FlynnToxicology, 2020
Dual Mechanisms Suppress Meloxicam Bioactivation Relative to SudoxicamDustyn A. Barnette, and Noah R.) FlynnToxicology, 2020Thiazoles are biologically active aromatic heterocyclic rings occurring frequently in natural products and drugs. These molecules undergo typically harmless elimination; however, a hepatotoxic response can occur due to multistep bioactivation of the thiazole to generate a reactive thioamide. A basis for those differences in outcomes remains unknown. A textbook example is the high hepatotoxicity observed for sudoxicam in contrast to the relative safe use and marketability of meloxicam, which differs in structure from sudoxicam by the addition of a single methyl group. Both drugs undergo bioactivation, but meloxicam exhibits an additional detoxification pathway due to hydroxylation of the methyl group. We hypothesized that thiazole bioactivation efficiency is similar between sudoxicam and meloxicam due to the methyl group being a weak electron donator, and thus, the relevance of bioactivation depends on the competing detoxification pathway. For a rapid analysis, we modeled epoxidation of sudoxicam derivatives to investigate the impact of substituents on thiazole bioactivation. As expected, electron donating groups increased the likelihood for epoxidation with a minimal effect for the methyl group, but model predictions did not extrapolate well among all types of substituents. Through analytical methods, we measured steady-state kinetics for metabolic bioactivation of sudoxicam and meloxicam by human liver microsomes. Sudoxicam bioactivation was 6-fold more efficient than that for meloxicam, yet meloxicam showed a 6-fold higher efficiency of detoxification than bioactivation. Overall, sudoxicam bioactivation was 15-fold more likely than meloxicam considering all metabolic clearance pathways. Kinetic differences likely arise from different enzymes catalyzing respective metabolic pathways based on phenotyping studies. Rather than simply providing an alternative detoxification pathway, the meloxicam methyl group suppressed the bioactivation reaction. These findings indicate the impact of thiazole substituents on bioactivation is more complex than previously thought and likely contributes to the unpredictability of their toxic potential.
@article{barnette2020meloxicam_dual, title = {Dual Mechanisms Suppress Meloxicam Bioactivation Relative to Sudoxicam}, author = {Barnette, Dustyn A. and others (including Flynn, Noah R.)}, year = {2020}, journal = {Toxicology}, }
2019
- Biochem. Pharm.
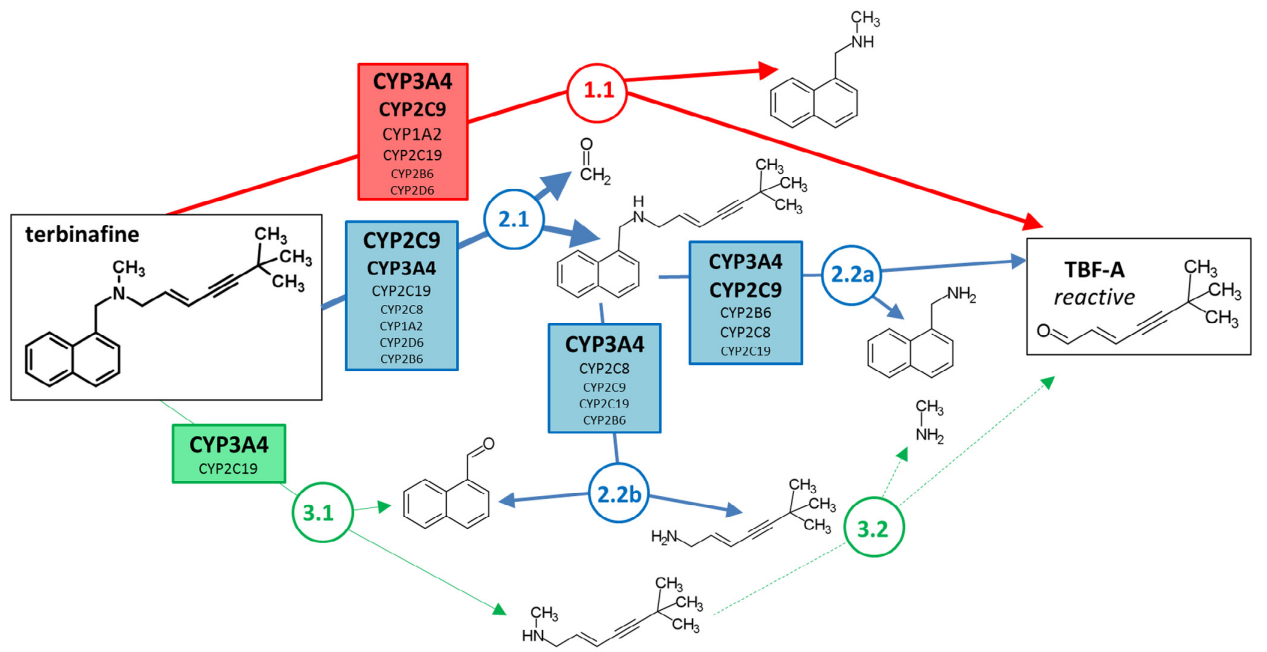 Comprehensive Kinetic and Modeling Analyses Revealed CYP2C9 and 3A4 Determine Terbinafine Metabolic Clearance and BioactivationDustyn A. Barnette, Mary A. Davis, Noah R. Flynn, and 1 more authorBiochemical Pharmacology, 2019
Comprehensive Kinetic and Modeling Analyses Revealed CYP2C9 and 3A4 Determine Terbinafine Metabolic Clearance and BioactivationDustyn A. Barnette, Mary A. Davis, Noah R. Flynn, and 1 more authorBiochemical Pharmacology, 2019Terbinafine N-dealkylation pathways result in formation of 6,6-dimethyl-2-hepten-4-ynal (TBF-A), a reactive allylic aldehyde, that may initiate idiosyncratic drug-induced liver toxicity. Previously, we reported on the importance of CYP2C19 and 3A4 as major contributors to TBF-A formation. In this study, we expanded on those efforts to assess individual contributions of CYP1A2, 2B6, 2C8, 2C9, and 2D6 in terbinafine metabolism. The combined knowledge gained from these studies allowed us to scale the relative roles of the P450 isozymes in hepatic clearance of terbinafine including pathways leading to TBF-A, and hence, provide a foundation for assessing their significance in terbinafine-induced hepatotoxicity. We used in vitro terbinafine reactions with recombinant P450s to measure kinetics for multiple metabolic pathways and calculated contributions of all individual P450 isozymes to in vivo hepatic clearance for the average human adult. The findings confirmed that CYP3A4 was a major contributor (at least 30% total metabolism) to all three of the possible N-dealkylation pathways; however, CYP2C9, and not CYP2C19, played a critical role in terbinafine metabolism and even exceeded CYP3A4 contributions for terbinafine N-demethylation. A combination of their metabolic capacities accounted for at least 80% of the conversion of terbinafine to TBF-A, while CYP1A2, 2B6, 2C8, and 2D6 made minor contributions. Computational approaches provide a more rapid, less resource-intensive strategy for assessing metabolism, and thus, we additionally predicted terbinafine metabolism using deep neural network models for individual P450 isozymes. Cytochrome P450 isozyme models accurately predicted the likelihood for terbinafine N-demethylation, but overestimated the likelihood for a minor N-denaphthylation pathway. Moreover, the models were not able to differentiate the varying roles of the individual P450 isozymes for specific reactions with this particular drug. Taken together, the significance of CYP2C9 and 3A4 and to a lesser extent, CYP2C19, in terbinafine metabolism is consistent with reported drug interactions. This finding suggests that variations in individual P450 contributions due to other factors like polymorphisms may similarly contribute to terbinafine-related adverse health outcomes. Nevertheless, the impact of their metabolic capacities on formation of reactive TBF-A and consequent idiosyncratic hepatotoxicity will be mitigated by competing detoxification pathways, TBF-A decay, and TBF-A adduction to glutathione that remain understudied.
@article{barnette2019terbinafine, title = {Comprehensive Kinetic and Modeling Analyses Revealed {CYP2C9} and {3A4} Determine Terbinafine Metabolic Clearance and Bioactivation}, author = {Barnette, Dustyn A. and Davis, Mary A. and Flynn, Noah R. and others}, year = {2019}, journal = {Biochemical Pharmacology}, } - Chem. Res. Tox.
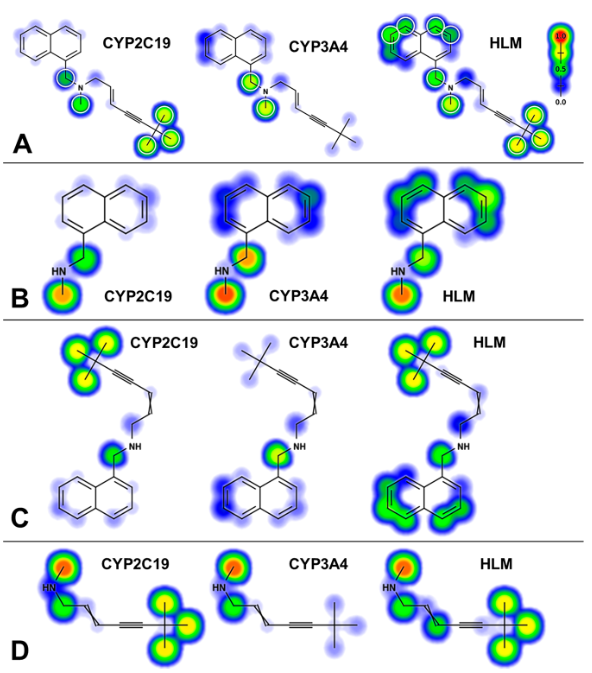 CYP2C19 and 3A4 Dominate Metabolic Clearance and Bioactivation of Terbinafine Based on Computational and Experimental ApproachesMary A. Davis, Dustyn A. Barnette, Noah R. Flynn, and 1 more authorChemical Research in Toxicology, 2019
CYP2C19 and 3A4 Dominate Metabolic Clearance and Bioactivation of Terbinafine Based on Computational and Experimental ApproachesMary A. Davis, Dustyn A. Barnette, Noah R. Flynn, and 1 more authorChemical Research in Toxicology, 2019Lamisil (terbinafine) is an effective, widely prescribed antifungal drug that causes rare idiosyncratic hepatotoxicity. The proposed toxic mechanism involves a reactive metabolite, 6,6-dimethyl-2-hepten-4-ynal (TBF-A), formed through three N-dealkylation pathways. We were the first to characterize them using in vitro studies with human liver microsomes and modeling approaches, yet knowledge of the individual enzymes catalyzing reactions remained unknown. Herein, we employed experimental and computational tools to assess terbinafine metabolism by specific cytochrome P450 isozymes. In vitro inhibitor phenotyping studies revealed six isozymes were involved in one or more N-dealkylation pathways. CYP2C19 and 3A4 contributed to all pathways, and so, we targeted them for steady-state analyses with recombinant isozymes. N-Dealkylation yielding TBF-A directly was catalyzed by CYP2C19 and 3A4 similarly. Nevertheless, CYP2C19 was more efficient than CYP3A4 at N-demethylation and other steps leading to TBF-A. Unlike microsomal reactions, N-denaphthylation was surprisingly efficient for CYP2C19 and 3A4, which was validated by controls. CYP2C19 was the most efficient among all reactions. Nonetheless, CYP3A4 was more selective at steps leading to TBF-A, making it more effective in terbinafine bioactivation based on metabolic split ratios for competing pathways. Model predictions did not extrapolate to quantitative kinetic constants, yet some results for CYP3A4 and CYP2C19 agreed qualitatively with preferred reaction steps and pathways. Clinical data on drug interactions support the CYP3A4 role in terbinafine metabolism, while CYP2C19 remains understudied. Taken together, knowledge of P450s responsible for terbinafine metabolism and TBF-A formation provides a foundation for investigating and mitigating the impact of P450 variations in toxic risks posed to patients.
@article{davis2019terbinafine, title = {{CYP2C19} and {3A4} Dominate Metabolic Clearance and Bioactivation of Terbinafine Based on Computational and Experimental Approaches}, author = {Davis, Mary A. and Barnette, Dustyn A. and Flynn, Noah R. and others}, year = {2019}, journal = {Chemical Research in Toxicology}, }